Financing led by existing investors Fountain Healthcare Partners and Panakès Partners
Neuromod will use funds to accelerate commercialisation in the USA and Europe.
Neuromod Devices Ltd. (Neuromod), an Irish medical device company specialising in tinnitus, has successfully closed a €10 million equity financing to expand the availability of its tinnitus treatment device, Lenire.
Oversubscribed Financing to Drive Commercialisation
Neuromod has raised €10 million of equity in an expansion of its Series B fundraisings. The financing was oversubscribed and was led by existing investors Fountain Healthcare Partners and Panakès Partners, backing Neuromod’s mission to advance tinnitus care for patients globally.
Neuromod has been making Lenire available through audiology and ENT practices throughout the USA and Europe. Proceeds from the financing will be used to meet demand for Lenire through sustainable commercial expansion in the USA and Europe and expand on existing opportunities in the US Department of Veteran Affairs (USVA).
Following FDA approval in March 2023, more than 100 clinics throughout the USA now treat tinnitus patients with Lenire. Availability of Lenire has also expanded in Europe with clinics in 14 countries now using the device. In the last 6 months, the number of clinics in the UK trained to use Lenire has doubled, and it is available to patients in Sweden for the first time.
In June 2024, Neuromod was awarded a Federal Supply Schedule 65 II Medical Equipment and Supply Contract from the US Government, making Lenire a treatment option for the 2.9 million US Veterans living with tinnitus [v] through the USVA.
35 USVA facilities have been trained to provide treatment with Lenire with more scheduled for training in 2025.
Real-World Evidence – Substantial Momentum
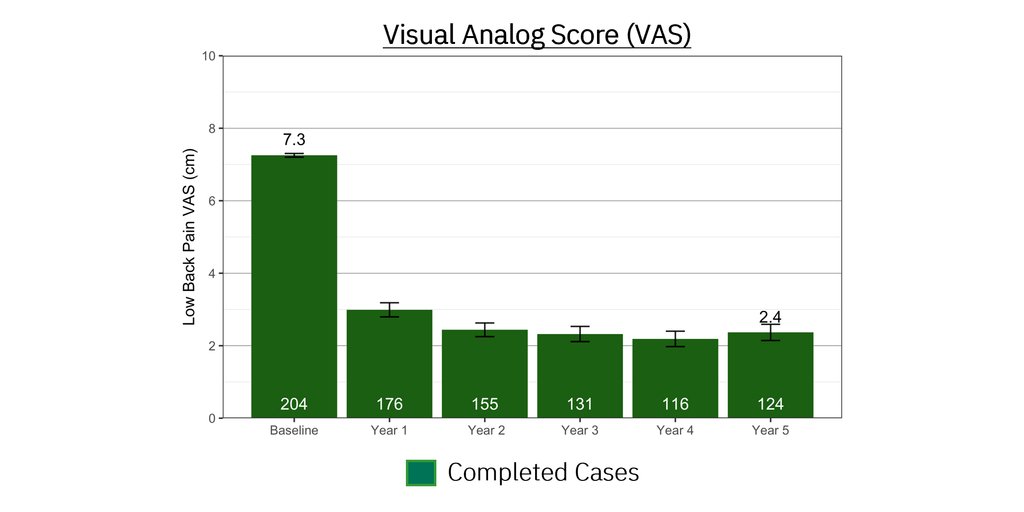
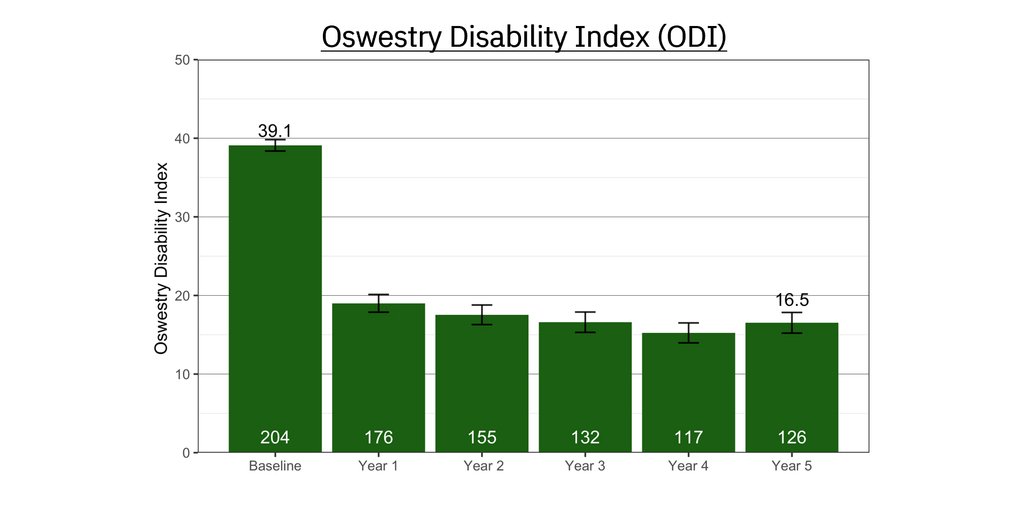
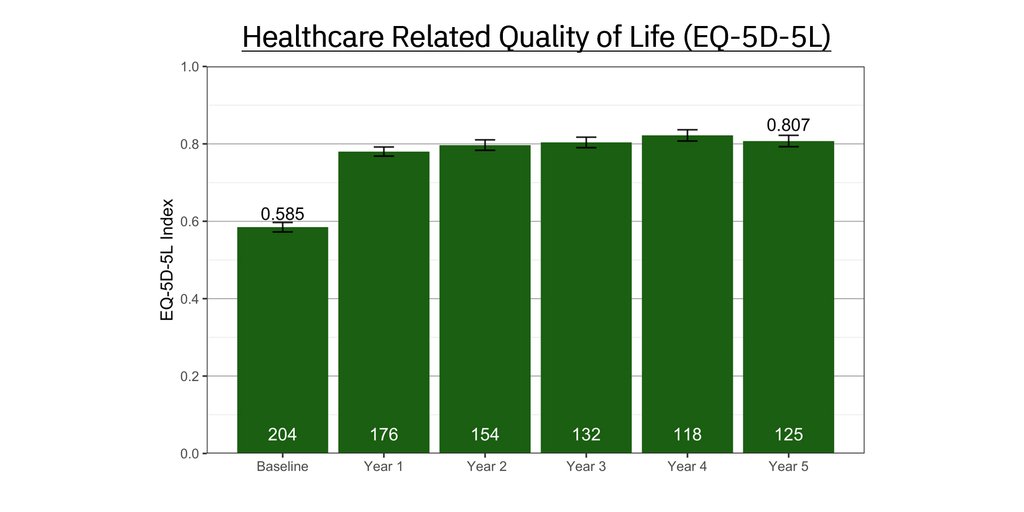
Positive results for tinnitus patients treated with Lenire in real-world settings at independent USA-based clinics have been compiled with a base of over 1,500 patients that continues to grow. In what will be the first of a series of planned real-world evidence publications, results from Alaska Hearing & Tinnitus Center showed that 91.5% of 220 patients reported clinically significant improvement in their tinnitus [vi]. This data is consistent with, and in many instances outperforms, data from Lenire’s large-scale clinical trials.
These results followed the publication of Lenire’s pivotal controlled clinical trial results, which led to US FDA approval and featured as the cover-story in peer-reviewed journal, Nature Communications [iv]. This article is in the 99th percentile of more than 250,000 tracked Nature articles.
Neuromod Closes €10m Financing Comments
Commenting on the news, Dr. Ross O’Neill PhD, Founder & CEO of Neuromod said “We are delighted to announce an oversubscribed financing at a pivotal time when we are driving forward with our mission of making Neuromod the category creator for tinnitus globally.”
“Tinnitus is the largest unmet need in hearing healthcare globally and is the number one service-connected disability among US veterans and military personnel. I am proud of the progress Neuromod is making to deliver our market-surpassing treatment to as many tinnitus patients as possible while enabling care providers’ expertise to be commercially rewarded. I am also grateful for the continued support of our investors who share our vision of advancing tinnitus care globally.” Dr. O’Neill continued.
Dr. Manus Rogan, Chairman of Neuromod and Managing Partner of Fountain Healthcare Partners commented, “Recent results from tinnitus patients using Lenire in the real-world show that it represents a new standard of care for tinnitus. The successful closing of this financing ensures more patients will get access to this standard of care as quickly as possible.”
Alessio Beverina, Managing Partner of Panakès Partners said, “Panakès is pleased with the progress of Neuromod since our investment, with significant clinical trial, FDA approval, real-world evidence, and commercial success in both Europe and the USA; and it is proud to continue supporting Neuromod’s work to bring a new standard of care to a historically underserved patient population.”
Emily E. McMahan, Owner of Alaska Hearing and Tinnitus Center and author of the clinic’s Real World Evidence Paper said, “Impressive clinical trial results for Lenire led me to early adoption of the landmark tinnitus treatment technology.”
“In my clinic, and my colleagues’ clinics, we are seeing results that are superior to clinical trial results.” Dr. McMahan continued.
About Neuromod Devices Ltd
Founded in 2010, Neuromod Devices Ltd. is a medical technology company headquartered in Dublin, Ireland. Neuromod specialises in the design, development, and commercialisation of neuromodulation technologies to address the clinical needs of underserved patient populations who live with chronic and debilitating conditions. The lead application of Neuromod’s technology is in the field of tinnitus, where Neuromod has completed extensive clinical trials to confirm the efficacy of its non-invasive neuromodulation platform in this common disorder. For more information visit www.neuromoddevices.com.
About Tinnitus
Tinnitus, commonly known as ‘ringing in the ears’, is a complex neurological condition that causes a perception of sound when there is no external source. Tinnitus affects an estimated 15% of the global adult population.
The management of tinnitus poses significant burden on healthcare systems. A 2021 study estimated the socioeconomic costs of tinnitus in Germany at €21.9 billion per annum. In the USA it’s estimated the Veterans Benefits Administration paid out approximately $5.8 billion through its Veterans Compensation benefits program for tinnitus in 2023.
The American Tinnitus Association, the leading advocacy body in the USA for people living with the condition, has recently revised its estimate that 50 million Americans live with tinnitus upward to 70 million.
About Lenire
Lenire is the first non-invasive bimodal neuromodulation tinnitus treatment device shown to soothe and relieve tinnitus in a large-scale clinical trial. Lenire works by delivering mild electrical pulses to the tongue, through an intra-oral component called the ‘Tonguetip®’, combined with auditory stimulation through headphones. This combination drives changes in the brain to treat tinnitus. To date, the device has been used in large-scale clinical trials with over 700 patients.
Lenire has CE-mark certification for the treatment of tinnitus under the supervision of an appropriately qualified healthcare professional in Europe and has received a De Novo grant of approval by the US FDA. Further details about Lenire including a list of providers can be found at www.lenire.com.
Connect with Neuromod
LinkedIn: linkedin.com/neuromod
X: x.com/NeuromodDevices
Facebook: facebook.com/neuromoddevices/
References & Notes
(i) Baguely et al., Tinnitus, The Lancet (2013), sciencedirect.com/science/article/pii/S0140673613601427
(ii) Conlon et al., Sci. Transl. Med. 12, eabb2830 (2020)
(iii) Conlon et al., Different bimodal neuromodulation settings reduce tinnitus symptoms in a large randomized trial, Sci Rep, doi.org/10.1038/s41598-022-13875-x (2022)
(iv) Boedts M, B. A., Khoo G, et al. Combining sound with tongue stimulation for the treatment of tinnitus: a controlled pivotal trial. Nature communications (2024)
(v) US VA Benefits Report Fiscal Year 2023: https://www.benefits.va.gov/REPORTS/abr/
(vi) McMahan, E.E. and Lim, H.H., 2024. Effectiveness of bimodal neuromodulation for tinnitus treatment in a real-world clinical setting in United States: A retrospective chart review. medRxiv., pp.2024-08; doi: https://doi.org/10.1101/2024.08.22.24312175 [preprint]
(vii) Tziridis K, Friedrich J, Brüeggemann P, Mazurek B, Schulze H. Estimation of Tinnitus-Related Socioeconomic Costs in Germany. Int J Environ Res Public Health. 2022 Aug 22;19(16):10455. doi: 10.3390/ijerph191610455. PMID: 36012089; PMCID: PMC9407899.
(viii) https://www.linkedin.com/posts/patrickalynch_tinnitus-activity-7270503831304654848-VbWN/